Piattaforma di Informatica Molecolare
Description
The Molecular Informatics group mainly deals with the identification and optimization of biologically active molecules through the use of in silico techniques. The approaches used range over classical molecular modeling techniques for virtual screening to the combination of modern chemoinformatics in house developed tools. Over the years the team has developed various experiences in the field of medicinal chemistry and computational chemistry. The expertise acquired by team members is synergistically exploited for the creation of molecular libraries, creating and validating reliable theoretical models to be used for subsequent virtual screening of ligands (VLS). The outcome of the models generated and optimised are further validated experimentally through biological or biophysical tests. The molecular informatics group is also involved in the exploration and enrichment of the chemical space to create the most suitable molecular libraries to be used for biological screening campaigns. In recent years, the collaboration with different academic groups from university of Palermo and Vienna allowed the development of approaches based on the use of deep learning for the activity prediction of small molecules. Furthermore, during the last year, the group has set the in silico Biologics platform that will be used for the design and optimisation of biological drugs.
Expertise
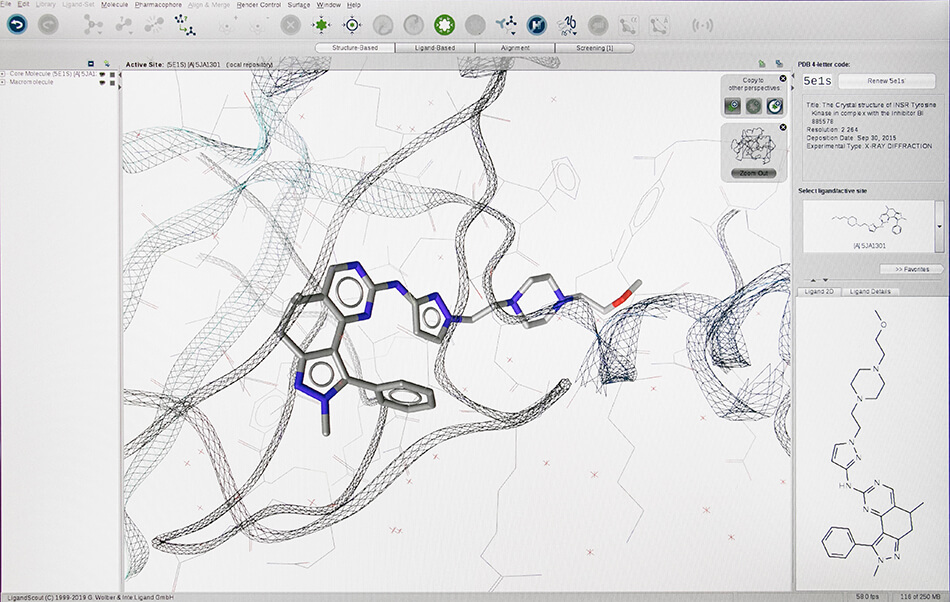
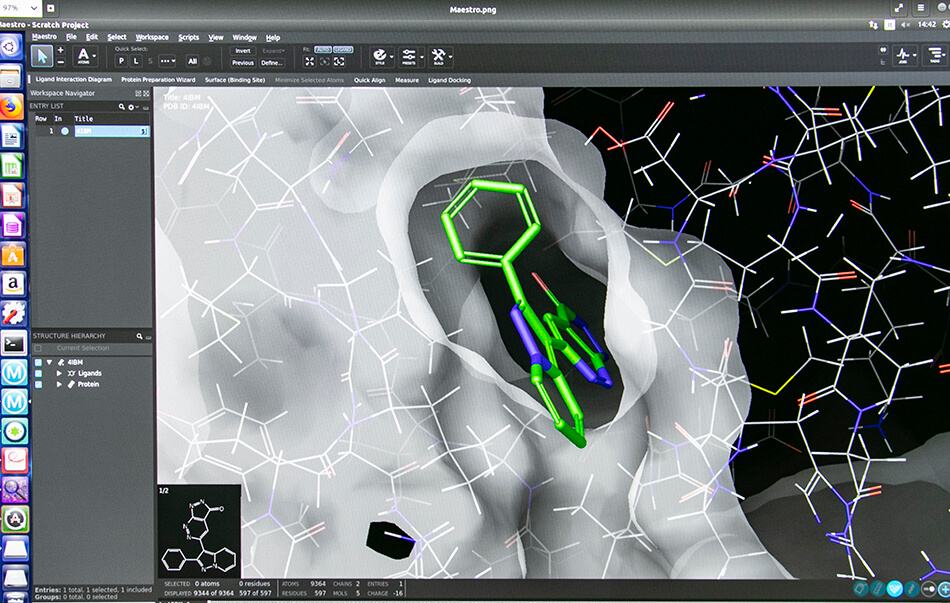
– Structure based virtual screening (Docking and Pharmacophore)
– Ligand Based virtual screening (pharmacophore, molecular descriptors based models, QSAR and 3D QSAR)
– Molecular Dynamics
– Dynamic pharmacophore (hybrid technique based on the use of pharmacophores from the molecular dynamics trajectory)
– Chemical Database creation and management
– Chemical data mining
– Machine Intelligence in Drug Design
– Biologics design
Facilities & Equipment
Software
Software
- Schrödinger suite for small molecule drug discovery
- Schrödinger suite for biologics drug discovery
- LigandScout expert suite
- Autodock and Autodock Vina
- AlvaDesc/AlvaModel
- DESMOND (OPLS2005 and OPLS3e, OPLS4)
- AMBER
- NAMD
- VMD
- GROMACS
- KNIME
integrated in silico platform
The group has participated to the creation of an integrated platform (OBIND) for molecular network analysis in collaboration with the Bioinformatics group
Contacts:
Collaboration:
- University of Vienna (Pharmaceutical Chemistry Dep.), Austria
- Universitè de Paris Cité, France
- Università di Napoli Federico II, Italy
- Univeristà degli Studi di Verona, Italy
- Consiglio Nazionale delle Ricerche IFT-CNR, Palermo, Italy
- Univeristà degli Studi di Palermo, Italy
Hardware
Hardware
- 6 Workstations
- Server: 80 cores e 2 x NVIDIA Tesla K80
- Server: 96 cores e 2 x NVIDIA A100
Calculation capability:
-
- Library optimisation ∼ 6,000 molecules/min
- Virtual Screening HTVS ∼ 5,000molecules/min
- Virtual screening SP ∼ 1,500 molecules/min
- Molecular Dynamics ∼ 200 ns/day/Card (on 40,000 atoms system)

Publications