
Sviluppo di algoritmi per modellizzare il network di interazione di microRNA e relativi target
Abstract
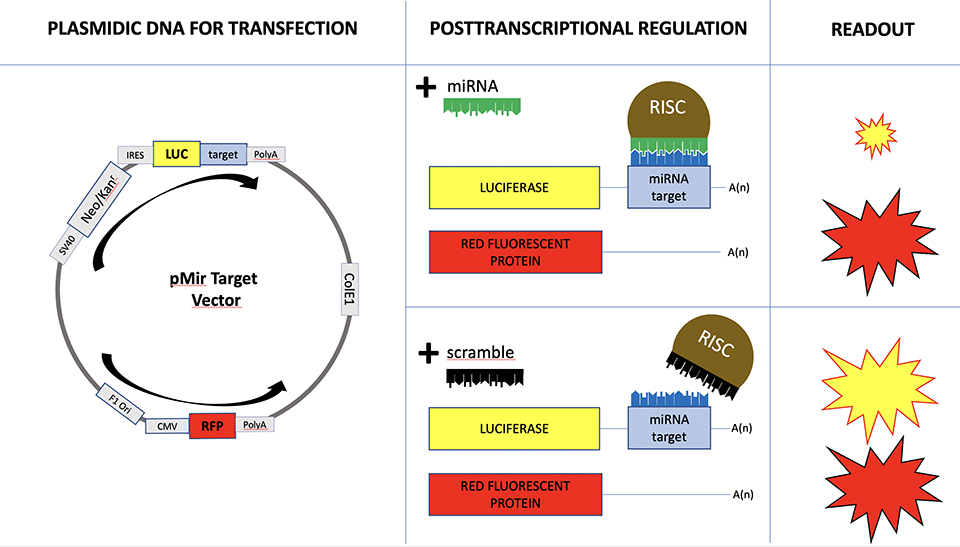
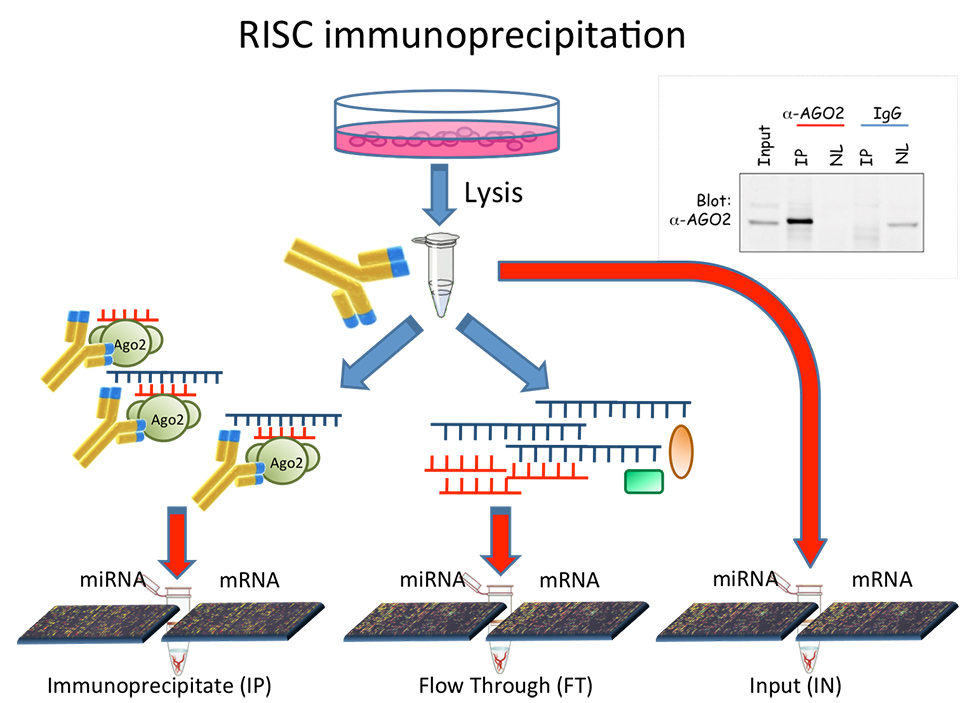
I microRNA sono piccoli filamenti di RNA con importanti funzioni regolatorie dei loro target, tra i quali compaiono gli RNA messaggeri (mRNA) codificanti per proteine. Ad oggi sono stati riconosciuti circa 2.000 microRNA e ciascuno di essi regola l’espressione di migliaia di target. Poiché il genoma umano comprende circa 20.000 geni codificanti per proteine, ci troviamo di fronte a una fitta e complessa rete di interazioni, chiamato mirnoma. Lo scenario è in più complicato dal fatto che ciascun tessuto cellulare possiede uno specifico profilo di espressione genica, per cui la rete di interazioni presente è diversa in ciascun tessuto. In questo progetto, ci proponiamo di modellizzare le reti di interazione, focalizzando lo studio su tessuti tumorali per individuare le anomalie sulla rete di interazioni rispetto ai rispettivi tessuti sani. I profili di espressione di microRNA e mRNA, utili a modellizzare la rete di interazione specifica di un tessuto, possono essere ricavati attraverso tecniche high throughput data analysis, come ad esempio le tecnologie basate su microarray o NGS. Con le suddette tecnologie è possibile misurare simultaneamente i livelli di espressione dei microRNA e mRNA presenti nel tessuto in analisi. Sarà obiettivo dei nostri algoritmi sfruttare tali dati per modellizzare e confrontare le reti di interazione tra microRNA e loro target in tessuti diversi.
Impatto:
Ricchissime sono oggi le banche dati che rendono pubblicamente accessibili Big Data biologici. Infatti, è apprezzata e incentivata la condivisione dei dati utilizzati per ottenere i risultati descritti nelle riviste scientifiche internazionali. Ad esempio, i profili di espressione genica, sono depositati in banche dati come Gene Expression Omnibus o ArrayExpress. Chiunque sia interessato ad un particolare tessuto cellulare avrà già a disposizione un corredo di profili di espressione da studi già pubblicati. Tali dati offrono informazione sull’espressione dell’intero genoma nei tessuti di interesse e vengono soprattutto utilizzati per eseguire un primo screening e individuare su quali marcatori focalizzare la futura ricerca. A fronte di una enorme mole di dati disponibili, ciò che manca sono algoritmi di analisi di dati in grado integrare più fonti di Big Data. Mentre sono ormai consolidati algoritmi utili ad individuare i geni o i microRNA differenzialmente espressi tra due tipologie di tessuto cellulare, utili per individuare le anomalie nei profili di espressione di microRNA e di mRNA, non esiste ad oggi alcun metodo per individuare quali anomalie si ripercuotono sulle interazioni tra microRNA e i mRNA. Lo sviluppo di tali metodi porterà nuovi strumenti per la comprensione delle cause di patologie come il cancro, arricchendo la ricerca di “quali sono i geni” con l’informazione su “quali sono le interazioni” coinvolte nella patologia sotto esame.
Pipeline
-
CLINICAL
NEED -
DISEASES
ANALYSIS - DISCOVERY
-
PRECLINICAL
VALIDATION -
PRECLINICAL
DEVELOPMENT -
CLINICAL
STUDIES
Principal Investigator
Contatto
Aree terapeutiche:
Prodotto:
Biomarcatori – farmaci biologici
Collaborazioni:
- Istituto per la Ricerca e l’Innovazione Biomedica (IRIB) – CNR, Palermo, Italia
- Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche (STEBICEF) – UNIPA, Palermo, Italia
- Dipartimento di Scienze Economiche, Aziendali e Statistiche (SEAS) – UNIPA, Palermo, Italia
- Dipartimento di Biologia Computazionale (CSB) – University of Pittsburgh, Pittsburgh, Stati Uniti
Scarica il pdf del progetto